
医療機器等の許認可について

医薬品医療機器等法では、医療機器および体外診断用医薬品(以下医療機器等)
を製造若しくは海外から輸入することにより、国内の市場に出荷して、流通する過
程を、その役割・機能に応じて、製造販売業(いわゆる元売)、製造業、修理業、
販売・貸与業等に区分しています。業としてこれらの事業を行おうとする者は、法
の定める許認可手続きを経る必要があります。
<目次>
1.医療機器等の製造販売業ついて
2.医療機器等の製造販売業の許可要件
3.医療機器等の製造販売の承認・認証・届出
4.医療機器等の製造業について
5.医療機器等の製造業の登録要件
6.医療機器等の修理業について
7.医療機器等の販売・貸与業について
1.医療機器等の製造販売業ついて
- (1)医療機器あるいは体外診断用医薬品とは(定義)
- 医療機器とは、人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は人若
しくは動物の身体の構造若しくは機能に影響を及ぼすことが目的とされている機械器具等(再生
医療等製品を除く。)であつて、政令で定めるものをいいます。
医療機器は、製造や販売などが医薬品医療機器等法で規制されており、どのようなものが「医
療機器」に該当するか具体的に定められています。メスやピンセットのような小物類から、コン
タクトレンズ、救急絆創膏、体温計など身近なもの、体内に植込む治療用の心臓ペースメーカ、
CT、MRI、放射線治療装置等の大型機器類まで広範囲にわたります。
(医薬品医療機器等法第2条第4項より)
- また、体外診断用医薬品とは、専ら疾病の診断に使用されることが目的とされている医薬品の
うち、人又は動物の身体に直接使用されることのないものをいい、疾病の診断に使用する医薬品
で身体に直接使用しないで、血液・尿便・唾液などを検査するために使用する試薬のこと。
(医薬品医療機器等法第2条第14項より)
- (2)医療機器等の製造販売業について
- 「製造販売業」という許可制度は、平成17年4月1日から、新たに導入されました。
「製造販売業」には、品質管理監督システム(QMS)に基づいて、製品を出荷判定して市場
に流通させる役割・機能があります。製品のライフサイクル上における、市場出荷前の「製造業
」と市場出荷後の「販売業」や「修理業」とを関連付けるとともに、最終消費者の製品の使用ま
でをつなぐ役割・機能があり、いわゆる元売り行為を行います。製造販売業では、製造を行うこ
とができません。
- 製造販売業者は、その取扱う製品についての市場に対する責任者であって、自社で製造する場
合のほかに、他社に製造を委託している場合や、海外で製造し輸入する場合についても、その品
質を確保する責任があります。
製造販売業者は、消費者からの情報や、使用している成分等の情報を広く収集・評価して、製
品の安全性を常に確保するとともに、必要な場合には、健康被害の発生を未然に防ぎ、拡大を防
止して消費者を保護するために、製品の回収を行います。
- <医療機器の製造販売の手続き全般の関係図>
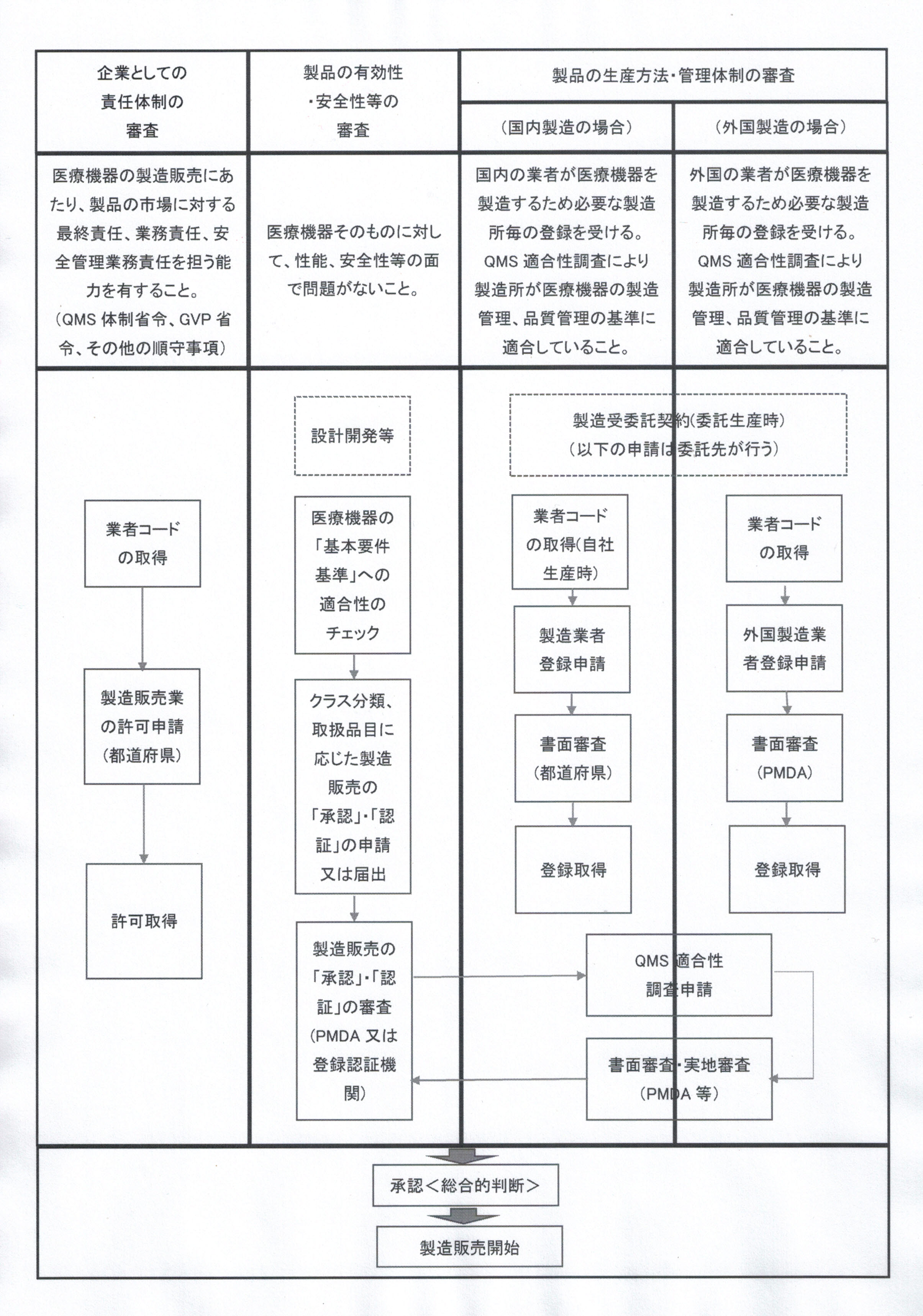
- ※参考:PMDA「医療機器の製造販売手順」
- (3)製造販売業の許可
- 医療機器等の製造販売を業として行う場合には、医療機器のクラス分類又は医療機器等の種類
に応じ、それぞれ厚生労働大臣の「許可」を受けなければなりません。(下表参照)
なお、医療機器等の製造販売業の許可は、5年ごとにその更新を受けル必要があります。
- <医療機器のクラス分類又は体外診断用医薬品の種類に応じた許可の種類>
-
医療機器のクラス分類又は体外診断用医薬品の種類 許可の種類 高度管理医療機器(クラスⅢ及びⅣ) 第一種医療機器製造販売業許可 管理医療機器(クラスⅡ) 第二種医療機器製造販売業許可 一般医療機器(クラスⅠ) 第三種医療機器製造販売業許可 体外診断用医薬品 体外診断用医薬品製造販売業許可 - (医薬品医療機器等法第23条の2より)
- (4)製造販売の承認・認証・届出
- 医薬品等の「製造販売」を行う場合には、上記の業の許可に加えて、医療機器のクラス分類又
は体外診断用医薬品の種類より、取り扱い品目に応じて、承認・認証・届出の申請手続きを経る
必要があります。取り扱い品目に応じて、製造販売について承認・認証を取得する又は届け出る
場合には、下表のとおり、医療機器のクラス分類又は体外診断用医薬品の種類に応じて、
QMS適合性調査を受けることにより行われますます。
- <医療機器のクラス分類又は体外診断用医薬品の種類に応じた承認・認証・届出>
-
医療機器のクラス分類又は体外診断用医薬品の種類 承認・認証・届出の種類 ・高度管理医療機器(クラスⅢ及びⅣ)のうち指定高度管理医療
機器でないもの
・管理医療機器(クラスⅡ)のうち指定管理医療機器でないもの
・認証、届出品目以外の体外診断用医薬品製造販売承認 ・指定高度管理医療機器(クラスⅢ及びⅣ)及び指定管理医療機
器(クラスⅡ)
・法23条の2の23第1項の規定により厚生労働大臣が基準を定
めて指する体外診断用医薬品製造販売認証 ・一般医療機器(クラスⅠ)
・法23条の2の5第1項の規定により厚生労働大臣が基準を定め
て 指定する体外診断用医薬品製造販売届出 - (医薬品医療機器等法第23条の2の5ほかより)
- (5)医療機器等外国製造業者の登録ほか輸入手続きについて
- 外国において我が国に輸出する医療機器等を製造しようとする者(以下、医療機器等外国製造
業者という)は、医薬品医療機器法施行規則第114条の8各号に掲げる製造工程に係る医薬品医
療機器法第23条の2の4による「登録」を受けていることが、当該医療機器等の製造販売承認の
要件となります。すなわち、医療機器等外国製造業者は、製造工程のうち、設計、組立て、滅菌
その他厚生労働省で定めるものを行う製造所において、品目に応じた「医療機器等外国製造業者
の登録」を取得する必要があります。外国製造業者の登録権者は厚生労働大臣となります。
なお、医療機器等外国製造業者が、我が国に輸出する医療機器等を国内で製造販売させるため
に承認を申請することができますが、その場合には、当該承認に係る医療機器による保健衛生上
の危害の発生の防止に必要な措置を採らせるために、あらかじめ許可を受けた製造販売業者を選
任してその承認申請を行わなければなりません。これに対して、厚生労働大臣は品目ごとに承認
を与えることができます。なお、登録申請に先立ち、当該外国製造業者及び製造所の業者コード
の登録が必要です。また、平成28年1月1日より輸入届が廃止されました。
(医薬品医療機器等法第23条の2の4、同第23条の2の5、同第23条の2の 20ほかより)
- 医薬品医療機器等法以外には、電動・電熱式医療機器について、電気用品安全法により、電気
用品輸入事業届出書の届け出義務があります。届出をした事業者(届出事業者)が輸入する電気
用品は、経済産業省の定める規格基準に適合していることが必要です。
また、登録認証機関による指定管理医療機器の認証に際して、工業標準化法によるJIS規格へ
の適合が求められることがあります。
2.医療機器等の製造販売業の許可要件
- 医療機器等の製造販売業許可取得の要件は、以下の2つです。
- <2-1> 体制省令・GVP省令への適合について
- <2-2> 人的要件を含むその他の順守事項
- また、許認可等の申請手続きの要領は、以下を参照ください。
<2-3> 医療機器等の製造販売業許可の申請手続き要領
<2-1> 体制省令・GVP省令への適合について
- (1)体制省令について
- 製造販売業者は、体制省令に適合する製造管理又は品質管理業務体制の確保が必要です。
体制省令とは、「医療機器又は体外診断用医薬品の製造管理又は品質管理に係る業務を行う体
制の基準に関する省令(平成26年厚生労働省令第94号)」(全4条)で、QMS省令(医療機器及
び体外診断用医薬品の製造管理及び品質管理の基準に関する省令)を遵守するために必要な体制
を定めたものです。医療機器又は体外診断用医薬品の製造販売業の許可要件として制定された厚
生労働省令です。製品を製造する製造所の製造管理及び品質管理に対する、製造販売業者として
の管理体制もその内容に含まれます。
体制省令の要点は、以下の(ア)組織の体制の整備、(イ)適切な人員の配置の2つです。
- (ア)組織体制の整備について
- 医療機器等製造販売業者はQMS省令を遵守するための体制を整備する必要があります。
具体的には以下の要求事項を満たす必要があります。
○品質管理監督システムの確立。(QMS省令第5条)
○品質管理監督システムの文書化及び実施並びにその実効性の維持。(QMS省令第6条、第7
条)
ただし、品質方針・品質目標等の作成については、限定第三種医療機器製造販売業を除く。
○品質管理監督文書の管理。(QMS省令第8条、第67条)
○品質管理監督記録の管理。(QMS省令第9条、第68条)(体制省令第3条第1項)
- (注1)限定第三種医療機器製造販売業者とは
一般医療機器のうち製造管理又は品質管理に注意を要するものとして厚生労働大臣が指定す
る医療機器以外の医療機器(限定一般医療機器)のみを製造販売する製造販売業者のことです
限定一般医療機器に対するQMS省令の適用範囲について、設計開発等の適用されない条文、
一部適用されに条文があります。
(注2)限定一般医療機器とは
一般医療機器には、QMS省令(医療機器及び体外診断用医薬品の製造管理及び品質管理の
基準に関する省令)第6条第1項の規定に基づき製造管理又は品質管理に注意を要するものと
して厚生労働大臣が指定する医療機器と、それ以外の限定一般医療機器があります。すべての
医療機器等にはQMS省令が適用されることが原則ですが、限定一般医療機器に対するQMS省
令の適用範囲については、設計開発等の適用されない条文、一部適用されない条文があります
- (イ)適切な人員の配置について
- 医療機器等製造販売業者はQMS省令の規定を遵守するために、それぞれの資格要件に応じ
た以下の人員の配置を適切に行う必要があります。
- (a)管理監督者(QMS省令第2条第16項)
製造販売業者等の品質管理監督システムに係る業務を最上位で監督する役員等。
(b)管理責任者。(限定第三種医療機器製造販売業者を除く)(QMS省令第16条)
製造販売業者等の役員、管理職の地位にある者その他これに相当する者。
(c)医療機器等総括製造販売責任者。(医薬品医療機器等法施行規則第114条の49)
(d)国内品質業務運営責任者。(QMS省令第72条第1項)
以下の要件を満たす者。
・製造販売業者における品質保証部門の責任者。
・品質管理業務その他これに類する業務の従事経験。(3年以上)
・国内の品質管理業務を適正かつ円滑に遂行しうる能力を有すること。
・医療機器等の販売に係る部門に属する者でないことその他国内の品質管理業務の適正か
つ円滑な遂行に支障を及ぼすおそれがないこと。
(体制省令第3条第2項を参照)
- (2)GVP省令について
- 製造販売業者は、製造販売している製品について、安全性を確保するために、法(GVP省令)
に定める基準に従って、製造販売後安全管理に関する業務を行う必要があります。
- 「医薬品、医薬部外品、化粧品、医療機器及び再生医療等製品の製造販売後安全管理の基準に
関する省令(平成16年厚生労働省令第135号)」(全5章16条)という、医薬品(原薬を除く)
等の製造販売後安全管理に関する基準で、製造 販売業の許可要件として制定された厚生労働省
令です。「GVP(Good Vigilance Practice)省令」と略称します。
- 医薬品等の製造販売をするにあたり、製造販売後に必要となる安全管理として、以下のような
安全管理情報(医薬品等の品質・有効性・安全性に関する事項、その他医薬品等の適正な使用の
ための情報)の収集、検討、手順書等に基づく安全確保措置の実施手順について規定しています
(ア)総括製造販売責任者の設置と業務。
(イ)安全管理責任者の業務(安全確保業務の統括等)。
(ウ)安全管理情報の収集業務(副作用・有害作用など安全性に関する情報を消費者や販売店、
医療関係者、業界団体、行政・外国等からの収集ルール)。
(エ)情報の検討と安全確保措置の立案業務(表示や使用上の注意の変更、国への報告や回収・
廃棄等の検討・立案ルール)。
(オ)安全確保措置の実施(表示や使用上の注意の変更、国への報告や回収・廃棄等の対応措置
の実施ルール)。
(カ)文書や記録の管理(保存方法や保存年限、改訂履歴等のルール)。
(キ)品質部門と安全部門あるいは営業部門など各部門との連携と他部門からの独立等。
<2-2> 人的要件・その他の順守事項
- (1)人的要件
- (ア)申請者の欠格要件
- 申請者(法人であるときは、医療機器の製造販売に関する業務を行う役員)は、医薬品医療
機器等法第23条の2の2の第3号に定める申請者の欠格要件である、下記(a)から(f)に該
当しないこと。
(a)法第75条第1項の規定により許可を取り消され、取り消しの日から3年を経過していな
い者。
(b)法第75条の2第1項の規定により登録を取り消され、取り消しの日から3年を経過してい
ない者。
(c)禁錮以上の刑に処せられ、その執行を終わり、又は執行を受けることがなくなった後、
3年を経過していない者。
(d)(a)から(c)までに該当する者を除くほか、この法律、麻薬及び向精神薬取締法、
毒物及び劇物取締法その他薬事に関する法令で政令で定めるもの又はこれに基づく処分に
違反し、その違反行為があった日から2年を経過していない者。
(e)成年被後見人又は麻薬、大麻、あへん若しくは覚醒剤の中毒者。
(f)精神の機能の障がいにより医療機器製造業の業務を適正に行うにあたって必要な認知、
判断及び意思疎通を適切に行うことができない者。
(医薬品医療機器等法第23条の2の2の第3号、同第5条第3号準用より)
- (イ)総括製造販売責任者の設置・資格要件
- 医療機器等の製造販売業者は、体制省令、QMS省令、GVP省令で求める業務を遂行するた
めに総括製造販売責任者を設置しなければなりません。
(医薬品医療機器等法第23条の2の14)
- <総括製造販売責任者の資格要件>
- 医薬品医療機器等法第23条の2の14に規定する医療機器等の製造版業の総括製造販売責任者
は、次の各号のいずれかに該当する者でなければなりません。
- (A)高度管理医療機器又は管理医療機器
(第一種、第二種の医療機器製造販売業)
- (a)大学等で物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、医学
又は歯学に関する専門の課程を修了した者。
(b)旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情
報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した後、
医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務に
3年以上従事した者。
(c)医薬品、医療機器又は再生医療等製品の品質管理又は製造販売後安全管理に関する業務
に五年以上従事した後、別に厚生労働省令で定めるところにより厚生労働大臣の登録を受
けた者が行う講習を終了した者。 - (e)厚生労働大臣が前三号に掲げる者と同等以上の知識経験を有すると認めた者。
(医薬品医療機器等法施行規則第114条の49第1項第1号から第4号より)
- (B)一般医療機器(第三種の医療機器製造販売業)
- (a)旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情
報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した者。
(b)旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、情
報学、金属学、電気学、機械学、薬学、医学又は歯学に関する科目を修得した後、医薬品
医薬部外品、化粧品、医療機器又は再生医療等製品の品質管理又は製造販売後、安全管理
に関する業務に三年以上従事した者。
(c)厚生労働大臣が前二号に掲げる者と同等以上の知識経験を有すると認めた者。
(医薬品医療機器等法施行規則第114条の49第2項第1号から第3号より)
- (C)体外診断用医薬品
- (a)薬剤師。(医薬品医療機器等法第23条の2の14第1項より)
- (ウ)安全管理業務に係る組織、責任者について
- 医療機器等の製造販売後安全管理に係るGVPでの要求事項は、製造販売業の許可の種類(第
一種・第二種・第三種)により異なり、第二種・第三種ではそれぞれ除外されている規定があ
ります。
- (A)第一種製造販売業組織(組織、責任者)
- (a)第一種製造販売業者は、次に掲げる要件を満たす安全確保業務の統括に係る部門(以下
「安全管理 統括部門」という。)を置かなければなりません。
(α)総括製造販売責任者の監督下にあること。
(β)安全確保業務を適正かつ円滑に遂行しうる能力を有する人員を十分に有すること。
(γ)医薬品等の販売に係る部門その他安全確保業務の適正かつ円滑な遂行に支障を及ぼすお
それのある部門から独立していること。
(b)第一種製造販売業者は、次に掲げる要件を満たす安全確保業務の責任者(以下、「安全
管理責任者」という。)を置かなければなりません。
(α)安全管理統括部門の責任者であること。
(β)安全確保業務その他これに類する業務に三年以上従事した者であること。
(γ)安全確保業務を適正かつ円滑に遂行しうる能力を有する者であること。
(δ)医薬品等の販売に係る部門に属する者でないことその他安全確保業務の適正かつ円滑な
遂行に支障を及ぼすおそれがない者であること。
また、医薬品医療機器等法第114条の59各号に掲げるものの全部又は一部を安全管理責
任者以外の者に行わせる場合にあっては、当該業務を適正かつ円滑に遂行しうる能力を有
する当該業務の実施に係る責任者(安全管理実施責任者)を置かなければなりません。
(GVP省令第4条より)
- (B)第二種、第三種製造販売業者
- (a)第二種製造販売業者は、次に掲げる要件を満たす安全管理責任者を置かなければなりま
せん。
(α)安全確保業務を適正かつ円滑に遂行しうる能力を有する者であること。
(β)医薬品等の販売に係る部門に属する者でないことその他安全確保業務の適正かつ円滑な
遂行に支障を及ぼすおそれがない者であること。また、安全確保業務を行う部門は医薬品
等の販売に係る部門その他安全確保業務の適正かつ円滑な遂行に支障を及ぼすおそれのあ
る部門から独立していなければなりません。
(GVP省令第13条、第15条より)
- (2)表示等について
- (ア)法定表示について
- 医療機器は、その医療機器又はその直接の容器若しくは直接の被包に、体外診断用医薬品は
その直接の容器又は直接の被包に、医薬品医療機器等法で定められた事項を表示しなければな
りません。
(医療機器:医薬品医療機器等法第63条、体外診断用医薬品:医薬品医療機器等法第50条よ
り)
- (イ)添付文書等の記載について
- 医療機器等には、これに添付する文書又はその容器若しくは被包に、最新の論文その他によ
り得られた知見に基づき、法令で定められた事項を記載しなければなりません。
(医療機器:医薬品医療機器等法第63条の2、体外診断用医薬品:医薬品医療機器等法第52条
より)
- (ウ)添付文書等記載事項等の届出について
- 特定高度管理医療機器(クラスⅣの高度管理医療機器)については製造販売開始時及び変更
の際は、あらかじめ、当該医療機器の添付文書等記載事項のうち、使用及び取扱い上の必要な
注意等を厚生労働大臣に届け出なければなりません。
(医薬品医療機器等法第63条の3より)
- (エ)添付文書等に記載してはならない事項について
- 医療機器等は、これに添付する文書、その医療機器等又はその容器もしくは被包に記載して
はならない事項が、医薬品医療機器等法により下記のように定められています。
-医薬品医療機器等法第54条(医療機器は法第64条で準用)【一部抜粋】-
・当該医療機器等に関し虚偽又は誤解を招くおそれのある事項
・承認を受けていない効能、効果又は性能
・保健衛生上危険がある用法、用量又は使用期間
- (3)回収(改修)について
- 製造販売業者が自ら製造販売した医療機器について、保健衛生上の被害の発生又は拡大の防止
のため、製品の市場からの回収が必要となることがあります。その場合には、迅速かつ適切に対
応する必要があります。そのため、日ごろからQMS・GVP等の手順書及び管理体制を整備し、
関連部署との連携体制 を整えておくことが重要になります。回収(改修)に着手したときは速
やかに監督官庁に報告しなけ ればなりません。
(医薬品医療機器等法第68条の11、同規則第228条の22より)
- (ア)回収処理の流れ
- (A)情報の入手
- QMS上の製品の品質等に関する情報及び、GVP上の安全管理情報などを入手。
- (B)情報の分析
- 情報の種類を整理して内容を分析。
- (C)回収の必要性の判断
- 分析結果及び関連法令や通知等に基づき、回収の必要性についての判断
(考慮するべきポイント)
(a)有効性及び安全性の観点からの判断
(b)医薬品医療機器等法違反又は承認事項からの逸脱
(c)不具合範囲の特定に関する判断
(d)混入した異物の種類と製品の性質の判断
- (D)回収の決定
- (E)報告
- 監督官庁(都道府県の報告先)への速やかな報告。
(医薬品医療機器等法第68条の11を参照)
- (F)「回収の概要」掲載・回収着手報告書・報道発表
- (a)クラス分類:安全性と有効性の両者を勘案し、総合的な「健康被害」としてクラス分類
を行う。
基本的にクラス2と考える。健康被害発生の可能性及び重篤度のバランスにより、クラス
1、クラス3となる。
・クラス1 その製品の使用等が重篤な健康被害又は死亡の原因となり得る場合
・クラス3 その製品の使用等が健康被害の原因とはまず考えられない場合
(b)回収範囲:回収漏れによる保健衛生上の問題の拡大を防ぐため、不具合品を確実にカバ
ーできるような範囲を設定。(対象ロット、数量)
(c)危惧される具体的な健康被害:どんな健康被害が発生したか(時、場所、件数)、また
は発生しうるか、納入医療機関や、製造元との連携を密にして情報を集め、検討。
- (G)納入施設への情報提供・回収措置
- QMS手順書等の回収処理に関する手順に基づいて、関係部門との連携のもと、回収作業
- (H)回収終了
- 回収対象の回収が漏れなく終わったことを確認。
- (I)回収終了報告書
- 「回収終了報告書」(回収結果と再発防止策として特に自社内で講じた措置)を作成し、 監督官庁(都道府県の報告先)への速やかな報告。
- (4)許可証の掲示
- 製造販売業の許可証を主たる機能を有する事務所(総括製造販売責任者が在籍している事務所 )の見やすい場所に掲示してください。
(規則第114条の85第1項で準用する同規則第3条をより) - (補足)医療機器等の製造販売業の許可要件には、構造設備基準はありません。
<2-3>医療機器等の製造販売業許可の申請要領
(以下、東京都、兵庫県の参考事例を記載)
| 1.医療機器等製造販売業許可 申請書 |
|
|---|---|
| (1)提出先、申請先及び 問合せ先 |
<東京都庁> 健康安全研究センター 広域監視部 医療機器監視課医療 機器審査担当 〒169-0073 東京都新宿区百人町3-24-1 本館1階 電話03ー5937ー1044 <兵庫県庁> 健康福祉部 健康局 薬務課 薬務指導班 〒650ー8567 兵庫県神戸市中央区下山手通5丁目10番1号 電話078ー362ー3269 |
| (2)様式 |
FD申請ソフトによる。 |
| (3)受付期間、時間 |
月曜日から金曜日(祝日、年末年始を除く) 午前9時から午前11時30分まで。 |
| (4)提出部数 |
1部(申込控え 1部)、郵送不可。 |
| (5)手数料 |
第一種 146,200円 (現金) 第二種 128,500円 (現金) 第三種 92,900円 (現金) 体外診断用医薬品 128,500円 (現金) |
| (6)標準的事務処理期間 |
35日 (土日祝日、年末年始を除く) |
| 2.添付書類 |
|
| (ア)総括製造販売責任者の 雇契約書の写しまたは雇 用及び用関係を証する書 類 |
|
| (イ)総括製造販売責任者の 資を証する書類 <医療機器製造販売業の場 合> |
医薬品医療機器等法施行規則第114条の49第1項第1号 ・・・A 医薬品医療機器等法施行規則第114条の49第1項第2号 ・・・A、B 医薬品医療機器等法施行規則第114条の49第1項第3号 ・・・C 医薬品医療機器等法施行規則第114条の49第2項第1号 ・・・A 医薬品医療機器等法施行規則第114条の49第2項第2号 ・・・A、B A:「卒業証書」の写し(窓口での原本提示必須)又は 「卒業証明」(場合によって履修単位報告書必要) B:「従事年数証明書」 C:「講習会修了証」の写し(窓口での原本提示必須) |
| 同上<体外診断用医薬品製造 販業の場合> |
「薬剤師免許証」の写し(窓口での原本提示必須)。 |
| (ウ)案内図 |
最寄りの駅から事務所までの地図を添付。 |
| (エ)建物の配置図 |
同一地番内に複数の建物がある場合、フロアの一部に事務所 がある合は添付。 |
| (オ)平面図 |
許可の対象となる事務所の範囲がわかるもの。 |
| (カ)登記事項証明書 (登記簿謄本) |
申請日より6ヶ月以内に発行されたものを添付。 |
| (キ)役員の業務分掌表 |
登記された役員全員が「業務を行う役員」に該当する場合は 添付不要。 |
| (ク)申請者が欠格条項に 該当るか否かに関する 診断書 ・「業務を行う役員」 (注1)ついて、申請日 より3ヶ月以に診断され たものを添付。 ・「業務を行う役員」の うち、海外在住であり、 薬事に関する業務の意 思決定に直接関与して いないとみなされる役 員は、診断書の代わり に疎明書(注2)でも可。 |
注1:業務を行う役員:「法人の薬局等の業務を行う役員 の範囲について」(昭和57年3月薬企第19号)及び 「法人の薬局等の業務を行う役員の範囲について」の 一部改正について」 (平成18年5月薬食安発第525001号)を参照。 注2:疎明書:「疎明対象の役員が欠格条項に該当しないこ と」を本人、または法人としての代表者(代表取締役) が証明する文書。 規定の様式はありません。疎明書を提出する場合、 業務分掌表に当該役員について「海外在住であり、 薬事に関する意思決定に直接関与しない」旨を記載。 |
| (ケ)組織図 |
|
| (コ)製造管理又は品質管理 に係る業務を行う体制に 関する書類 |
国内品質業務運営責任者の資格要件を確認の上、作成。 |
| (サ)製造販売後安全管理に 係る体制に関する書類 |
安全管理責任者の資格要件を確認の上、作成。 |
| (シ)製造販売業の許可証の 写し |
申請者が現に製造販売業の許可を受けている場合は、取得 しているべての製造販売業許可証の写し。 |
| ※更新について |
医療機器等の製造販売業の許可は、5年ごとにその更新が 必要です |
3.医療機器等の製造販売の承認・認証・届出
- (1)医療機器等の製造販売の承認・認証・届出とは
- 医療機器等の製造販売を行うためには、業としての「製造販売業許可」とは別に、医療機器の
クラス分類を基に定められた手順に従い、医療機器その者に対して製造、安全性等の面で問題が
ないことについて、厚生労働省に申請し、品目ごとに「承認」・「認証」・「届出」の手続きを
経なければなりません。「承認」はPMDAにより、また「認証」は登録認証機関により、それぞ
れQMS適合性審査を受けることになります。
- (2)医療機器のクラス分類とQMS適合性調査について
- 医薬品医療機器等法では、人体に与えるリスクの程度に応じて医療機器をクラス分類し、この
分類に従って規制を変える仕組みを採用しています。日本の全ての医療機器は、いずれかの
JMDN(=日本の医療機器の一般的名称(注2):Japan Medical Device Nomenclature)に
該当します。
また、これらのJMDNには、「一般医療機器(クラスⅠ)」、「管理医療機器(クラスⅡ)」
「高度管理医療機器(クラスⅢ及びⅣ)」の医療機器のクラス分類(クラスⅠ~Ⅳ)が対応して
います。JMDNは、国際的な医療機器の一般的名称であるGMDN (Global Medical Device
Nomenclature) を積極的に取り入れたものです。また、医薬品医療機器等法の3つの分類
(「高度管理医療機器」、「管理医療機器」、「一般医療機器」)も、同様に、国際医療機器規
制整合化会議、日・米・欧・豪・加の5極(GHTF:Global Harmonization Task Force) にお
いて定められたクラス分類ルール(クラスⅠ~Ⅳ)を参考にして「一般医療機器(クラスⅠ)」
「管理医療機器(クラスⅡ)」、「高度管理医療機器(クラスⅢ及びⅣ)」として対応がなされ
ており、JMDN並びに医療機器のクラス分類ルールは、ともに国際整合化が図られています。
- <医療機器のクラス分類と審査(承認・認証・届出)>
(※)PMDA:独立行政法人 医薬品医療機器総合機構
-
クラ
ス
分類クラス分類の考え方 医薬品医
療機器等
法の分類医療機器
QMS適合
性調査製造販売
品目の承
認・認証
・届出基準
適合
性審査機
関(申
請先)Ⅳ 患者への侵襲性が高く、
不具合が生じた場合、生
命の危険に直結する恐れ
があるもの高度管理
医療機器必要 承認 - PMDA Ⅲ 不具合が生じた場合、人
体へのリスクが比較的高
いと考えられるもの同上 必要 承認 存在
しな
いPMDA 同上 同上 同上 同上 適合
しな
い同上 同上 指定高度
管理医療
機器同上 認証 適合
する登録認
証機関Ⅱ 不具合が生じた場合でも
、人体へのリスクが比較
的低いと考えられるもの管理医療機器 必要 承認 存在
しな
いPMDA 同上 同上 同上 同上 適合
しな
い同上 同上 指定管理
医療機器同上 認証 適合
する登録認
証機関Ⅰ 不具合が生じた場合でも
、人体へのリスクが極め
て低いと考えられるもの一般医療機器 不要 届出 - - - <体外診断用医薬品のクラス分類と審査(承認・認証・届出)>
-
クラ
ス
分類クラス分類の考え方 体外用診断
医薬品
QMS適合性
調査製造販売品
目の承認・
認証・届出基準
適合
性審査機
関Ⅲ 体外診断用医薬品を疾病の診断等
に使用した際、その診断情報リス
クが比較的大きく、情報の正確さ
が生命維持に与える影響が大きい
と考えられるもの。必要 承認 不適
合PMDA 同上 同上 同上 適合 同上 Ⅱ クラスⅢに該当しない体外診断用
医薬品のうち、一般用検査薬(
OTC)であるもの。必要 承認 不適
合PMDA 同上 同上 認証 適合 登録認
証機関Ⅰ クラスⅢに該当しない体外診断用
医薬品のうち、国内外で一般的な
ものとして認知されている較正用
標準物質が存在するのであって、
体外診断用医薬品の製造管理およ
び品質管理の一環として行われる
較正が比較的容易と認められ、か
つ、一般用検査薬(OTC)以外
のもの。必要 承認 不適
合PMDA 同上 不要 製造販売届
出適合 -
4.医療機器等の製造業について
- (1)製造業の登録
- 医療機器の製造とは、製造販売業者が自社で製造する場合のほかに、他社に製造を委託する場
合や、外国の製造所で製造する場合も含めて、医療機器・体外診断用医薬品(以下、医療機器等
という。)を製造することです。
- 医療機器等の製造を業として行う場合には、製造所(医療機器等の製造工程のうち、設計、組
立て、滅菌、その他の厚生労働省令で定める工程の作業を行うものに限る。)ごとに、厚生労働
省令で定めるところにより、厚生労働大臣の「登録」を受けなければなりません
全ての医療機器等にQMS省令が適用されるため、製造業者は、品質管理監督システム(
QMS)を基に施設や設備を管理する必要があります。製造業者は、製造販売業者が行う製造管理
及び品質管理に協力しなければなりません。
- 医療機器等の製造業者は、製造工程のうち、設計、組立て、滅菌、その他厚生労働省で定める
ものを行う製造所においては、品目に応じた「製造業登録」をする必要があります。医療機器等
の製造工程のうち、設計、組立て、滅菌、その他の厚生労働省令で定める工程については、製造
する品目の種類に応じて、下表のとおり規定されています。
- <製造業の登録が必要な製造工程(規則第114条の8)>
(○:要登録、✖:不要)
設計 主たる組立て
その他の主た
る製造工程滅菌 国内における
最終製品の保
管1.医療機器プログラム ○ ✖ ✖ ✖ 2.医療機器プログラムを
記録した記録媒体たる医
療機器○ ✖ ✖ ○ 3.一般医療機器 ✖ ○ ○ ○ 4.上記以外の医療機器 ○ ○ ○ ○ - <体外診断用医薬品の登録が必要な製造工程(規則第114条の8)>
(○:要登録、✖:不要)
設計 反応系に関与
する成分の最
終製品への充
填工程反応系に関与する
成分の最終製品へ
の充填工程以降の
すべての製造工程国内におけ
る最終製品
の保管1.放射性医薬品
である体外診断
用医薬品○ ○※ ○ ○※ 2.承認又は認証
が必要な体外診
断用医薬品○ ○ ✖ ○ 3.上記以外の体
外診断用医薬品✖ ○ ✖ ○ - ※「反応系に関与する成分の最終製品への充填工程以降のすべての製造工程」に含まれます。
- (2)外国の製造所で医療機器等を製造している場合の「外国製造業者登録」
- 外国において日本に輸出する医療機器等を製造しようとする者を、医薬品医療機器等法におけ
る医療機器等外国製造業者と言います。
全ての医療機器等にQMS省令が適用されるため、製造業者は、品質管理監督システム(
QMS)を基に施設や設備を管理する必要があります。外国製造業者もまた、製造販売業者が行う
製造管理及び品質管理に協力しなければなりません。
- 外国製造業者は、国内製造業者の登録と同様に、医薬品医療機器法施行規則第114条の8各号
に掲げる製造工程に係る医薬品医療機器法第23条の2の4による「登録」を受けていることが、
当該医療機器等の製造販売承認の要件となります。すなわち、医療機器等外国製造業者は、製造
工程のうち、設計、組立て、滅菌、その他厚生労働省で定めるものを行う製造所においては、品
目に応じた「医療機器等外国製造業者の登録」をする必要があります。
- 外国製造業者の登録権者は厚生労働大臣となります。なお、登録申請に先立ち、当該外国製造
業者及び製造所の業者コードの登録が必要です。
- 「1.(5)外国製造医療機器等の製造販売ほか輸入手続きについて」を参照。
5.医療機器等の製造業の登録要件
- 医療機器等の製造業登録の要件は、以下の2つです。
- <5-1> 製造管理及び品質管理の基準(QMS)の遵守
- <5-2> 人的要件・その他の順守事項
- また、許認可等の申請手続きの要領は、以下を参照ください。
<5ー3> 医療機器等の製造業登録の申請手続き要領
<5-1> 製造管理及び品質管理の基準(QMS)の遵守
- (1)製造販売業者が行う製造管理又は品質管理に協力
- 製造業者は、製造販売業者が行う製造管理又は品質管理に協力しなければなりません。
(医薬品医療機器等法23条の2の15第2項、医薬品医療機器等法施行規則第114条の58参照)
- (2)全ての医療機器等へのQMS省令の適用
- すべての医療機器及び体外診断用医薬品にQMS省令が適用されますので、製造販売業者及び
製造業者における製品の製造管理及び品質管理の方法をQMS省令に適合させる必要があります
(QMS省令第83条を参照)
なお、一般医療機器や承認を要しない体外診断用医薬品には、QMS省令の「設計開発」の規
定が適用されません。また、限定一般医療機器(一般医療機器のうち厚生労働大臣が指定した品
目以外のもの)を製造する製造所は、「設計開発」のほか、さらにQMS省令の一部の条項の適
用が除外されています。
- (3)製造業者の承認・認証のためのQMS適合性調査の実施
- 製造販売業者が品目の承認や認証を取得するためには、QMS適合性調査を受ける必要があり
ます。その際に、製造業者の調査も行われます。また、適合性調査は、承認等取得後の変更時お
よび5年ごとに定期的な調査も必要となります。
(医薬品医療機器等法第23条の2の5第2項第4号、第6項、第23条の2の23第2項第5号、第3項
を参照)
(注)QMS適合性調査の概要(クラス分類の概要、審査概要、審査機関(申請先)等)は、製
造販売業で記載する<医療機器のクラス分類と審査(承認・認証・届出)>、<体外診断用医
薬品のクラス分類と審査(承認・認証・届出)>を参照。
<5-2> 人的要件・その他の順守事項
- (1)人的要件
- (ア)申請者の欠格要件
- 申請者(法人であるときは、医療機器の製造に関する業務を行う役員)の欠格要件は、医薬
品医療機器等法第23条の2の3第4項(同第5条第3号を準用)により定めますが、その内容は
前記の医療機器等の製造販売業許可におけるものと同じです。
(「医療機器等の許認可 <2-2>(1)(ア)申請者の欠格要件」を参照。)
(医薬品医療機器等法第23条の2の3第4項、同第5条第3号準用より)
- (イ)責任技術者の設置
- 医療機器等製造業の製造所は、責任技術者を設置しなければなりません。
(医薬品医療機器等法第23条の2の14第3項)
- <責任技術者の資格要件>
- 医薬品医療機器等法第23条の2の14第3項に規定する化粧品の製造所の責任技術者は、次の
各号のいずれかに該当する者でなければなりません。
- (a)高度管理医療機器又は管理医療機器を製造する製造所
(α)大学等で、物理学、化学、生物学、工学、情報学、金属学、電気学、機械学、薬学、
医学又は歯学に関する専門の課程を修了した者。
(β)旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、
情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した
後、医療機器の製造に関する業務に3年以上従事した者。
(γ)医療機器の製造に関する業務に五年以上従事した後、別に厚生労働省令で定めるとこ
ろにより厚生労働大臣の登録を受けた者が行う講習を修了した者。
(δ)厚生労働大臣が前3号に掲げる者と同等以上の知識経験を有すると認めた者。
(医薬品医療機器等法施行規則第114条の53第1項第1号から第4号より)
- (b)一般医療機器のみを製造する製造所
(α)旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、
情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する専門の課程を修了した
者。
(β)旧制中学若しくは高校又はこれと同等以上の学校で、物理学、化学、生物学、工学、
情報学、金属学、電気学、機械学、薬学、医学又は歯学に関する科目を修得した後、医
療機器の製造に関する業務に三年以上従事した者。
(γ)厚生労働大臣が前2号に掲げる者と同等以上の知識経験を有すると認めた者。
(医薬品医療機器等法施行規則第114条の53第2項第1号から第3号より)
- (c)医療機器の製造工程のうち設計のみを行う製造所
(α)前2項に該当する者もしくは、製造業者が設計に係る部門の責任者として指定する者
(医薬品医療機器等法施行規則第114条の53第3項より)
- (d)体外診断用医薬品を製造する製造所
(α)薬剤師(医薬品医療機器等法第23条の2の14第5項より)
- (2)記録の作成
- 医療機器責任技術者又は体外診断用医薬品製造管理者は、製造及び試験に関する記録、その他
製造所の管理に関する記録を作成し、3年間(又は、有効期間+1年のうち長い期間)保管しなけ
ればなりません。ただし、薬事に関する他の法令等(次の項目で示す「QMS省令」等)で、記
録の作成及びその保管が義務づけられている場合は、それらにも従う必要があります。
(医薬品医療機器等法施行規則第114条の52、QMS省令第9条、同第68条より)
- (3)製造販売業者との取決め
- 製造販売業者は、QMS省令第72条の2により、製造業者との間で必要かつ十分な事項について
取り決め、文書化しなければなりません。そのため、製造業者は製造業務を開始する前に、製造
方法、試験方法等の変更や製品の品質等に関する情報の取扱などを製造販売業者と文書で取り決
めを行います。
(製造業者と製造販売業者が同一法人の場合は、社内規定等の文書に適切に規定します。)
- (4)許可証の掲示
- 製造業の登録証(原本)は、製造所の見やすい場所に掲示しなければなりません。
(医薬品医療機器等法施行規則第114条の85で準用する同規則第3条を参照)
<5-3> 医療機器等の製造業登録の申請要領
(以下、東京都、兵庫県の参考事例を記載)
| 1.医療機器等製造業登録 申請書 |
|
|---|---|
| (1)申請先及び問合せ先 |
<東京都庁> 健康安全研究センター 広域監視部 医療機器監視課 医療機器審査担当 〒169-0073 東京都新宿区百人町3-24-1 本館1階 電話 03-5937-1044 <兵庫県庁> 健康福祉部 健康局 薬務課 薬務指導班 〒650ー8567 兵庫県神戸市中央区下山手通5丁目10番1号 電話 078ー362ー3269 |
| (2)様式 |
FD申請ソフトによる |
| (3)受付期間、時間 |
月曜日から金曜日(祝日、年末年始を除く) 午前9時から午前11時30分まで |
| (4)提出部数 |
1部(申請者控え 1部)、郵送不可 |
| (5)手数料 |
38,200円(現金) |
| (6)標準的事務処理期間 |
20日 (土日祝日、年末年始を除く) |
| 2.添付書類 |
|
| (ア)登記事項証明書 (登記簿謄本) |
申請日から6ヶ月以内に発行されたものを添付。 |
| (イ)役員の業務分掌表 |
登記された役員全員が「業務を行う役員」に該当する場合は 添付不要。 |
| (ウ)申請者が欠格条項に該当 するか否かに関する診断書 もしは疎明書(注1) ※「業務を行う役員」(注2) について、申請日より3ヶ月 以内に診断もしくは疎明さ れたもを添付。 |
注1:「疎明対象の役員が欠格事項に該当しないこと」を本人、 または法人としての代表者(代表取締役)が証明する文 書 規定の様式はありません。 注2:「法人の薬局等業務を行う役員の範囲について」(昭和 57年3月薬企第19号)及び、「「法人の薬局等業務を行 う役員の範囲について」の一部改正について」 (平成18年5月薬食安発第525001号)を参照。 |
| (エ)責任技術者・製造管理者 の「雇用契約書の写し」又 は雇用及び使用関係を証す る書類」 |
|
| (オ)責任技術者・製造管理者 の資格を証する書類 <医療機器製造業> |
医薬品医療機器等法施行規則第114条の53第1項第1号…A 医薬品医療機器等法施行規則第114条の53第1項第2号…A,B 医薬品医療機器等法施行規則第114条の53第1項第3号…C 医薬品医療機器等法施行規則第114条の53第2項第1号…A 医薬品医療機器等法施行規則第114条の53第2項第2号…A,B 医薬品医療機器等法施行規則第114条の53第2項第3号…B 医薬品医療機器等法施行規則第114条の53第3項…D A 「卒業証書」の写し(窓口での原本提示が必須)又は「卒 業証明書」(学科等によって、単位履修(成績)証明書を 提出する場合があります。) B 「従事年数証明書」 C 「講習会修了証」の写し(窓口での原本提示必須) D なし |
| 同上 <体外診断用医薬品製造業> |
「薬剤師免許証」の写し(窓口での原本提示が必須) |
| (カ)製造所の場所を明らかに した図面 |
(カー1)案内図 最寄駅から製造所までの地図を添付。 (カー2)建物の配置図 同一地番内に複数の建物がある場合に添付。 建物内の一部に製造所がある場合は、フロア全体図も添付。 (カ-3)製造所の平面図登録対象となる製造所の範囲がわかる もの。 |
| (キ)許可証及び登録証の写し |
同一所在地において、他の区分の製造業の許可・登録を受けて いる場合は、当該製造業の許可・登録証の写し。 |
| ※更新について |
医療機器等の製造業の登録は、5年ごとにその更新が必要です。 |
6.医療機器等の修理業について
- (1)修理業とは
- 医療機器の修理とは、以下のように定義されているものを指しています。
医療機器の修理とは、故障、破損、劣化等の箇所を本来の状態・機能に復帰させること(当該
箇所の交換を含む。)をいうものであり、故障等の有無にかかわらず、解体の上点検し、必要に
応じて劣化部品の交換等を行うオーバーホールを含むものです。医療機器の修理を業として行お
うとする者は、事業所ごとに地方厚生局若しくは都道府県知事許可を得なければなりません。
- ただし、清掃、校正(キャリブレーション)、消耗部品の交換等の保守点検は修理に含まれな
いものであり、修理業の許可を必要としません。なお、修理業者を紹介する行為のみを行うにあ
っては修理業の許可は必要ないが、医療機器の修理業務の全部を他の修理業者等に委託すること
により実際の修理を行わない場合であっても、医療機関等から当該医療機器の修理の契約を行う
場合は、その修理契約を行った者は修理された医療機器の安全性等について責任を有するもので
あり、修理業の許可を要します。
- また、医療機器の仕様の変更のような改造は修理の範囲を超えるものであり、別途、医療機器
製造業の許可を取得する必要があります。
(平成17年3月 薬食機発第0331004号より)
- (2)修理業の許可
- 医療機器の修理を業として行う場合には、医療機器の修理区分に応じ、それぞれ厚生労働大臣
の「許可」を受けなければなりません。医療機器修理業の「許可」を取得するためには、修理を
行う事業所の構造設備を薬局等構造設備規則第5条に適合させる必要があります。
(薬局等構造設備規制第5条(医療機器の修理業の事業所の構造設備)を参照)
- 医療機器を修理する場合には、修理業者はあらかじめ医療機器製造販売業者に修理する医療機
器の品名、製造番号、故障内容を通知して、医療機器製造販売業者から修理指示を受けなければ
なりません。修理業者はその指示に基づいて修理を行ないます。
- なお、医療機器の修理業の許可は、5年ごとに更新を受ける必要があります。また、製造業者
が製造した品目をその製造所において修理する場合には、修理業許可は必要ありません。
(医薬品医療機器等法第40条の2を参照)
- (3)修理できる品目に応じた修理区分
- 医療機器を修理する場合には、修理できる品目に応じた修理区分ごとの「許可」が必要です。
医療機器の修理区分は、第1区分から第9区分の9つの区分に分かれています。更に、9つの区分
は「特定保守管理医療機器(特管)」と「特定保守管理医療機器以外の医療機器(非特管)」と
に分かれ、全18区分に分かれています。医療機器修理業者には、区分ごと、事業所ごとに「許
可」が与えられます。
- 特定保守管理医療機器は、医療機器のうち、保守点検、修理その他の管理に専門的な知識及び
技能を必要とすることからその適正な管理が行われなければ疾病の診断、治療又は予防に重大な
影響を与えるおそれがあるものとして、厚生労働大臣が薬事・食品衛生審議会の意見を聴いて指
定するものです。特定保守管理医療機器は、医療機器のクラス分類に関わらず、保守点検、修理
その他の管理に専門的な知識及び技能を必要とするものです。その内、設置にあたり組立が必要
でかつ組立の管理が必要な医療機器を、「設置管理医療機器」と言います。
- <薬事法施行規則別表2で示す修理区分の概要(平成17年3月薬食機発第0331004号より)>
-
特定保守管理医療機器(特管) 特定保守管理医療機器以外の医療機器
(非特管)特管第一区分:画像診断システム関連
非特管第一区分:画像診断システム関連 特管第二区分:生体現象計測・監視シス
テム関連非特管第二区分:生体現象計測・監視シス
テム関連特管第三区分:治療用・施設用機器関連 非特管第三区分:治療用・施設用機器関連 特管第四区分:人工臓器関連 非特管第四区分:人工臓器関連 特管第五区分:光学機器関連 非特管第五区分:光学機器関連 特管第六区分:理学療法用機器関連 非特管第六区分:理学療法用機器関連 特管第七区分:歯科用機器関連 非特管第七区分:歯科用機器関連 特管第八区分:検体検査用機器関連 非特管第八区分:検体検査用機器関連 特管第九区分:鋼製器具・家庭用医療
機器関連※非特管第九区分:鋼製器具・家庭用医療
機器関連※ - ※現在「特管第九区分」に該当する品目はありません。
- (4)医療機器等の修理業の許可の要件
- 医療機器等の修理業許可取得の要件は、以下の2つです。
(ア)薬局等構造設備規則への適合
(イ)人的要件(申請者の欠格要件、責任技術者を設置)
- (ア)薬局等構造設備規則への適合
- 医療機器修理業者は、医療機器の修理を行う事業所を薬局等構造設備規則第5条に適合させ
る必要があります。
(参考:薬局等構造設備規則第5条 抜粋)
医療機器の修理業の事業所の構造設備の基準は、次のとおりとする。
一 構成部品等及び修理を行った医療機器を衛生的かつ安全に保管するために必要な設備を有
すること。
二 修理を行う医療機器の種類に応じ、構成部品等及び修理を行った医療機器の試験検査に必
要な設備及び器具を備えていること。ただし、当該修理業者の他の試験検査設備又は他の試
験検査機関を利用して自己の責任において当該試験検査を行う場合であって、支障がないと
認められるときは、この限りでない。
三 修理を行うのに必要な設備及び器具を備えていること。
四 修理を行う場所は、次に定めるところに適合するものであること。
イ 採光、照明及び換気が適切であり、かつ、清潔であること。
ロ 常時居住する場所及び不潔な場所から明確に区別されていること。
ハ 作業を行うのに支障のない面積を有すること。
ニ 防じん、防湿、防虫及び防そのための設備を有すること。ただし、修理を行う医療機器
により支障がないと認められる場合は、この限りでない。
ホ 床は、板張り、コンクリート又はこれらに準ずるものであること。ただし、修理を行う
医療機器により作業の性質上やむを得ないと認められる場合は、この限りでない。
ヘ 廃水及び廃棄物の処理に要する設備又は器具を備えていること。
五 作業室内に備える作業台は、作業を円滑かつ適切に行うのに支障のないものであること。
- (イ)人的要件
- (A)申請者の欠格要件
- 医薬品医療機器等法第23条の2の3の第4号に定める、医療機器等の修理業許可に係る申請
者の欠格要件(同第5条第3号を準用)は、前記の医療機器等の製造販売業許可の申請者の
要件と同じす。
(「医療機器等の許認可 <2-2>(1)(ア)申請者の欠格要件」を参照。)
(医薬品医療機器等法第23条の2の3の第4号、同第5条第3号準用より)
- (B)責任技術者を設置する
- 修理業者は、医療機器の修理を実地に管理させるために、事業所ごとに医療機器修理責任
技術者を置かなければなりません。
(医薬品医療機器等法第40条の3より)
- <医療機器修理責任技術者資格要件>
- 医薬品医療機器等法第40条の3に規定する医療機器等の修理業の責任技術者は、次の各
号のいずれかに該当する者でなければなりません。
(a)特定保守管理医療機器の修理を行う修理業者。
(α)医薬品医療機器等法施行規則第188条第1項第1号イ
医療機器の修理に関する業務に3年以上従事した後、別に厚生労働省令で定めるところ
により厚生労働大臣の登録を受けた者が行う基礎講習及び専門講習を修了した者。
(β)医薬品医療機器等法施行規則第188条第1項第1号ロ
厚生労働大臣がイに掲げる者と同等以上の知識経験を有すると認めた者。
(第1区分、第2区分のみ存在)
- (b)特定保守管理医療機器以外の医療機器の修理を行う修理業者
(α)医薬品医療機器等法施行規則第188条第1項第2号イ
医療機器の修理に関する業務に三年以上従事した後、基礎講習を修了した者。
(β)医薬品医療機器等法施行規則第188条第1項第2号ロ
厚生労働大臣がイに掲げる者と同等以上の知識経験を有すると認めた者。
(現在、該当資格はありません。)
(5)<医療機器等の修理業の申請手続き要領>
(東京都、兵庫県の参考事例を記載)
| 1.医療機器修理業許可申請書 |
|
|---|---|
| (1)申請先及び問合せ先 |
<東京都庁> 健康安全研究センター 広域監視部 医療機器監視課 医療機器審査担当 〒169-0073東京都新宿区百人町3-24-1 本館1階 電話 03-5937-1044 <兵庫県庁> 健康福祉部 健康局 薬務課 薬務指導班 〒650-8567 兵庫県神戸市中央区下山手通5丁目10番1号 TEL 078-362-3269 |
| (2)様式 |
FD申請ソフトによる。 |
| (3)受付期間、時間 |
月曜日から金曜日(祝日、年末年始を除く) 午前9時から午前11時30分まで |
| (4)提出部数 |
提出部数1部 (申請者控え 1部)、郵送不可 |
| (5)手数料 |
75,200円 (現金) |
| (6)標準的事務処理期間 |
35日 (土日祝日、年末年始を除く) |
| 2.添付書類 |
|
| (ア)構造設備の概要一覧表 |
|
| (イ)案内図 |
最寄りの駅から事業所までの地図を添付。 |
| (ウ)建物の配置図 |
・同一地番内に複数の建物がある場合に添付。 ・建物内の一部に作業場所等がある場合はフロア全体図も添付。 |
| (エ)事業所の平面図 |
作業場所、保管場所及び試験場所等を明記し、寸法を記載。 |
| (オ)保管設備の詳細図 |
構成部品、未修理品、修理完了品の保管棚の立体図に寸法を 記載。 |
| (カ)修理用機械器具一覧表 |
|
| (キ)試験検査用機械器具一覧 表 |
|
| (ク)他の試験検査機関もしく は試験検査設備所有者との 契約書(写)あるいは利用 証明書 |
・他の試験検査設備又は、他の試験検査機関を利用する場合に 添付 ・転貸している物件を利用する場合は、建物所有者と転貸者間 の契 約書の写しも添付。 ※契約期間が許可有効期間を満たしていること。 ※医薬品医療機器等法により許可を有している製造業。 (登録のみの製造業は除く。) ・修理業を、他の試験検査設備もしくは他の試験検査機関とし て利用する場合は、構造設備概要一覧の備考欄に許可番号を 記載。 |
| (ケ)登記事項証明書 (登記簿謄本) |
申請日より6ヶ月以内に発行されたものを添付。 |
| (コ)役員の業務分掌表 |
登記された役員全員が「業務を行う役員」に該当する場合は 添付不要。 |
| (サ)申請者が欠格条項に該当 するか否かに関する診断書 もしくは疎明書(注1) (「業務を行う役員」 (注2)について、申請日 より3ヶ月以内に診断もしく は疎明されたものを添付。) |
注1:「疎明対象の役員が欠格事項に該当しないこと」を本人、 又は法人としての代表者(代表取締役)が証明する文書。 規定の様式はなし。 注2:「法人の薬局等の業務を行う役員の範囲について」(昭 和57年3薬企第19号)及び、「「法人の薬局等の業務を 行う役員の範囲について」の一部改正について」。 (平成18年5月薬食安発第525001号)を参照。) |
| (シ)責任技術者の「雇用契約 書の写し」又は「雇用及び 使用関係を証する書類」 |
|
| (ス)責任技術者の資格を証す る書類 |
(A)「基礎講習終了証」の写し(窓口での原本提示が必須)。 (B)「専門講習終了証」の写し(窓口での原本提示が必須)。 |
| ※更新について |
医療機器等の修理業の許可は、5年ごとにその更新が必要です。 |
7.医療機器等の販売・貸与業について
- (1)高度管理医療機器等の販売・貸与業の許可について
- 通常の医療機器を販売するためには、特に販売許可等を取得する必要はなく、小売店等で自由
に販売できます。
ただし、「高度管理医療機器」、並びに「管理医療機器」又は「一般医療機器」のうち「特定
保守管理医療機器」に該当する医療機器(高度管理医療機器等)について、業として、それらを
販売し、授与し若しくは貸与し、若しくは販売、授与若しくは貸与の目的で陳列し、又は管理医
療機器プログラム(管理医療機器のうちプログラムであるものをいう。)を電気通信回線を通じ
て提供すること。)を行うためには事前に許可の取得が必要です。
なお、上記の許可は、6年ごとにその更新を受ける必要があります。
- 許可の基準:以下の(ア)及び(イ)の要件を満たさなければなりません。
(医薬品医療機器等法第39条、第39条の2参照)
- (ア)営業所の構造設備が次の基準を満たしていること
- (A)採光、照明及び換気が適切であり、かつ、清潔であること。
(B)常時居住する場所及び不潔な場所から明確に区別されていること。
(C)取扱品目を衛生的に、かつ、安全に貯蔵するために必要な設備を有すること。
※(A)から(C)の規定は、医療機器プログラムの電気通信回線を通じた提供のみを行う
営業所については、適用しません。
- (イ)営業所に営業管理者(高度管理医療機器等営業管理者)を設置するこ と
- (A)指定視力補正用レンズ又はプログラム高度管理医療機器等のみを販売等す
る者以外の高度管理医療機器等販売業者
(a)医療機器の販売又は貸与に関する業務に3年以上従事した後、別に厚生労働省令で定め
るところにより厚生労働大臣の登録を受けた者が行う基礎講習を修了した者
(b)厚生労働大臣が上記(1)に掲げる者と同等以上の知識及び経験を有すると認めた者
・医師、歯科医師、薬剤師の資格を有する者
・医療機器の第一種製造販売業の総括製造販売責任者の要件を満たす者
・医療機器の製造業の責任技術者の要件を満たす者
・医療機器の修理業の責任技術者の要件を満たす者
・改正薬事法(平成18年法律第69号)附則第7条の規定により法第36条の4第1項に規定
する試験に合格したとみなされた者のうち、同条第2項の登録を受けた者
・財団法人医療機器センター及び日本医科器械商工団体連合会が共催で実施した医療機器
販売適正事業所認定制度「販売管理責任者講習」を修了した者
- (B)指定視力補正用レンズ等のみを販売等する高度管理医療機器等販売業者等
(a) 高度管理医療機器等(プログラム高度管理医療機器を除く。)の販売等に関する業務
に1年以上従事した後、別に厚生労働省令で定めるところにより厚生労働大臣の登録を受
けた者が行う基礎講習を修了した者
(b)非視力補正用コンタクトレンズの販売業及び貸与業に関する講習(販売業特別講習)を
修了した者
- (C)プログラム高度管理医療機器のみを販売提供等する高度管理医療機器等販
売業者等別に厚生労働省令で定めるところにより厚生労働大臣の登録を受け
た者が行う基礎講習を修了した者
- (D)指定視力補正用レンズ等及びプログラム高度管理医療機器のみを販売提供
等する高度管理医療機器販売業者等
上記(A)及び(C)参照
- (2)管理医療機器販売・貸与業の届出について
- 管理医療機器を販売・貸与するためには管理医療機器販売業(貸与業)の届出が必要です。
届出を行う際には、以下の(ア)及び(イ)の事項に御留意ください。
(医薬品医療機器等法第39条の3参照)
- (ア)届出の時点で、営業所の構造設備が次の基準を満たしていること
- (A)採光、照明及び換気が適切であり、かつ、清潔であること。
(B)常時居住する場所及び不潔な場所から明確に区別されていること。
(C)取扱品目を衛生的に、かつ、安全に貯蔵するために必要な設備を有すること。
※(A)から(C)の規定は、医療機器プログラムの電気通信回線を通じた提供のみを行う
営業所については、適用しません。
- (イ)届出の時点で、営業管理者(管理医療機器営業管理者)を設置するこ
と
- (A)特定管理医療機器(専ら家庭において使用される管理医療機器であって厚生労働
大臣の指定するもの以外の管理医療機器をいう。)の販売業者等
(a)規則第175条第1項前段該当者
(高度管理医療機器等の販売等に関する業務に1年以上若しくは特定管理医療機器の販売
等に関する業務(特定管理医療機器のうち補聴器、家庭用電気治療器若しくはプログラム
特定管理医療機器のみ、又は補聴器及び家庭用電気治療器のみ、補聴器及びプログラム特
定管理医療機器のみ、家庭用電気資料木及びプログラム特定管理医療機器のみ、補聴器、
家庭用電気治療器及びプログラム特定管理医療機器のみを販売等する業務を除く。)に
3年以上従事した後、別に厚生労働省令で定めるところにより厚生労働大臣の登録を受け
た者が行う基礎講習を修了した者)
(b)規則第175条第1項後段該当者
(前記(a)に掲げる者と同等以上の知識及び経験を有すると厚生労働大臣が認めた者)
・医師、歯科医師、薬剤師の資格を有する者
・医療機器の第一種製造販売業の総括製造販売責任者の要件を満たす者
・医療機器の製造業の責任技術者の要件を満たす者
・医療機器の修理業の責任技術者の要件を満たす者
・改正薬事法(平成18年法律第69号)附則第7条の規定により薬事法第36条の4第1項に
規定する試験に合格したとみなされた者のうち、同条第2項の登録を受けた者
・財団法人医療機器センター及び日本医科器械商工団体連合会が共催で実施した医療機器
販売適正事業所認定制度「販売管理責任者講習」を修了した者
・「検体測定室に関するガイドラインについて」(平成26年4月9日付医政発0409第4号
厚生労働省医政局長通知)別添で定める検体測定室の運営責任者である看護師又は臨床
検査技師(ただし、検体測定室における検査で使用される医療機器のみを販売等する営
業所に限る。)
- (B)特定管理医療機器のうち「補聴器」のみを販売等する販売業者等
(a)規則第175条第1項第1号前段該当者
(特定管理医療機器の販売等に関する業務(特定管理医療機器のうち家庭用電気治療器及
びプログラム特定管理医療機器のみを販売等する業務を除く。)に1年以上従事した後、
別に厚生労働省令で定めるところにより厚生労働大臣の登録を受けた者が行う基礎講習を
修了した者)
(b)規則第175条第1項第1号後段該当者
(前記(a)に掲げる者と同等以上の知識及び経験を有すると厚生労働大臣が認めた者)
上記(A)の(b)参照
- (C)特定管理医療機器のうち「家庭用電気治療器」のみを販売等する販売業者等
(a)規則第175条第1項第2号前段該当者
(特定管理医療機器の販売等に関する業務(特定管理医療機器のうち補聴器のみを販売
等する業務を除く。)に1年以上従事した後、別に厚生労働省令で定めるところにより厚
生労働大臣の登録を受けた者が行う基礎講習を修了した者)
(b)規則第175条第1項第2号後段該当者
(前記(a)に掲げる者と同等以上の知識及び経験を有すると厚生労働大臣が認めた者
)上記(A)の(b)参照
- (D)特定管理医療機器のうち「プログラム特定管理医療機器」のみを販売等する販売
業者等
(a)規則第175条第1項第3号前段該当者
(別に厚生労働省令で定めるところにより厚生労働大臣の登録を受けた者が行う基礎講
習を修了した者)
(b)規則第175条第1項第3号後段該当者
(前記(1)に掲げる者と同等以上の知識及び経験を有すると厚生労働大臣が認めた者)
上記(A)の(b)参照
- (E)特定管理医療機器のうち「補聴器及び家庭用電気治療器」のみを販売等する販売
業者等
上記(B)の(a)及び(C)の(a)参照。
- (F)特定管理医療機器のうち「補聴器及びプログラム特定管理医療機器」のみを販売
等する販売業者等
上記(B)の(a)及び(D)の(a)参照。
- (G)特定管理医療機器のうち「家庭用電気治療器及びプログラム特定管理医療機器」
のみを販売等する販売業者等
上記(C)の(a)及び(D)の(a)参照。
- (H)特定管理医療機器のうち「補聴器、家庭用電気治療器及びプログラム特定管理医療機
器」のみを販売等する販売業者等
上記(B)の(a)、(C)の(a)及び(D)の(a)参照。
- (I)特定管理医療機器以外の管理医療機器を販売等する販売業者等
- (医療機器の製造から使用まで終わり。)